
Photo by Edward Caldwell: James Spudich studies how mutations in cardiac myosin could play a role in a deadly heart disorder called hypertrophic cardiomyopathy.
Stanford Medicine News Center - March 20th, 2019 - by Bruce Goldman
Jim Spudich isn’t the kind of avid reader who devours a book in two days and moves immediately to the next on his nightstand. He just reads himself to sleep on evenings when he wants to reroute his brain from thinking about his work. Which is most of the time.
“Creative research is not a job — it’s a full-time, almost childlike obsession driven by the curiosity that all kids have,” said Spudich, PhD, professor of biochemistry. “I see myself as a kid who’s never had to grow up.”
Yet even scientists have to sleep. Spudich, 77, ordinarily gets his fair share — close to eight hours a night. But when he needs some help winding down, a good murder mystery will do it, especially if it’s set in the American Southwest, where he spent several years as a child and whose mesa-strewn terrain he’s flown over many times in the pilot’s seat of a small plane. (He’s a licensed pilot.)
Flying is the ultimate escape for Spudich, the Douglass M. and Nola Leishman Professor in Cardiovascular Disease. “I can go out once every couple of weeks for an hour, climb up into the clouds and get some perspective as everything down below disappears,” he said. In particular, his taste in bird’s-eye views favors mesas, which look like what you’d get if you sawed the top off a mountain.
One evening, in December 2014, Spudich’s wife of 54 years, Anna, who knows his tastes well, slipped him some bedtime reading, a murder mystery. Spudich knocked off a few chapters before falling asleep, only to awaken in the dark with the germ of a vision that would solve a mystery he’d been brooding over for more than a year.
“It’s not so unusual that we scientists go to bed dreaming about our research,” Spudich said. “What was different about this dream was that it changed the entire course of our work.”
Dying young
When he was 5, Spudich moved with his family from Illinois to Phoenix because his older sister had severe heart trouble, and her doctor had warned that she’d never make it through another cold winter. In the warm, dry Arizona climate, she survived for five more years before succumbing to a heart attack.
The heart beats because it’s made of muscle cells that rhythmically shrink in sync and then relax, pumping blood throughout the vascular system. Since 1969, Spudich has zeroed in on a pair of proteins that make all muscular contraction possible. His interest has led him to focus on a particular disorder called hypertrophic cardiomyopathy.
“HCM, as we often call it, was first recognized as a genetics-based disease only about 25 years ago,” Spudich said. “It’s epitomized by the phenomenon of young athletes in their prime keeling over from sudden death when nobody even knew anything was wrong with them.”
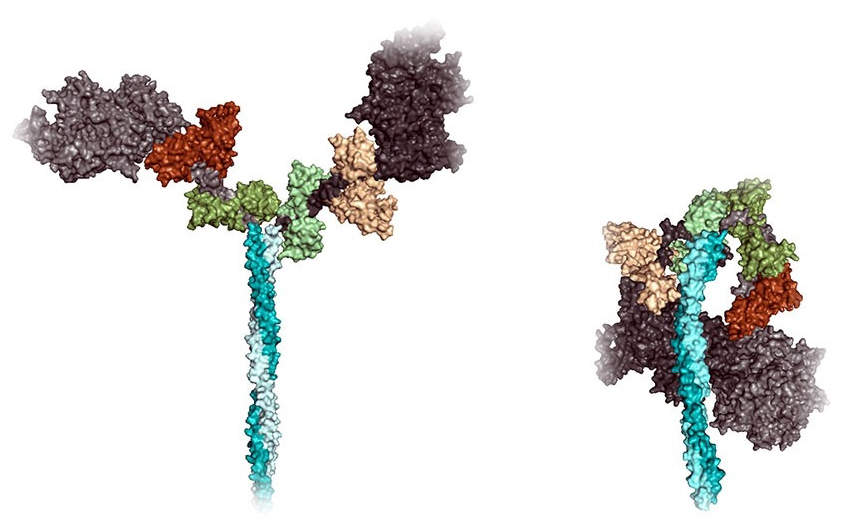
Graphic courtesy Jim Spudich: An illustration of a myosin
molecule in an “open” and “folded” position.
HCM’s defining clinical symptom is a hypercontractile heart. “It’s as if you’re out for a short run,” Spudich said. “The problem is, you’re doing that 24 hours a day, every day of your life.”
In response, the heart muscle thickens and eventually stiffens, choking off blood flow through the organ. The progression can end in sudden death.
One in every 500 people is affected by HCM. “The number of patients keeps getting bigger,” Spudich said. “We used to have no idea how many there were, because people weren’t checking.” But with the discovery that HCM could — and usually did — arise from genetic mutations, intensive gene screening has turned up hundreds of different mutations, mostly in a small number of the proteins of the muscle-contraction machinery, that can cause the disorder.
A riddle that has stymied Spudich for years is this: “We usually think of mutations as causing a protein not to work as well as the unmutated version does; they’re messing with what was a beautiful evolutionary design,” he said. “But HCM mutations are doing the opposite. They’re somehow causing the protein to work ‘better.’” The result is a heartbeat that’s too powerful.
More than a third of those mutations occur in the gene coding for a protein that’s intimately involved in every move we make and every beat of the heart. Spudich knows a thing or two about that protein, called myosin. He won the coveted Lasker Award in 2012, largely for inventing novel ways to study individual myosin molecules and their interactions with another protein called actin.
Of the 40 or so nearly identical versions of myosin produced in every cell of the body, Spudich pays the most attention to cardiac myosin, the version produced and used by heart muscle cells. In fact, Spudich’s eight-person lab is one of the few in the world bearing down on human cardiac myosin in a serious way, he said. “So, I have an obligation to keep working on this until we cross the finish line.”
Current HCM therapies leave much to be desired. Physicians rely on conventional heart drugs that, for example, slow the heartbeat. The ultimate treatment comes later: open-heart surgery to cut away excess heart muscle. “You can only do this once,” Spudich said.
Looking for clues
A myosin molecule’s general structure, known since the 1960s, resembles a two-headed monster: two large globular “heads” protruding from a stalklike tail. It’s long been known that myosin sometimes adopts a posture in which its heads fold over and snuggle up against its tail, reminiscent of a sleeping flamingo with its head tucked under a wing.
“But the relevance of this to HCM was essentially unknown,” Spudich said.
To see why it’s important, it helps to know how healthy muscles contract. Spudich has played a major role in explaining this in precise molecular detail.
A cardiac muscle cell contains perhaps a hundred repeating structural subunits called sarcomeres, arranged one after another in a series. A sarcomere is composed of myosin-rich “thick filaments” alternating with parallel “thin filaments” made of actin.

Courtesy James Spudich: An avid pilot, Spudich, shown here
in the mid-1990s, has flown over many mesas. He had a
dream about a mesa that provided an insight about what
may be at the root of hypertrophic cardiomyopathy.
Closer inspection reveals that each thick filament is knitted from the bottom halves of myosin molecules’ tails. In response to electrical impulses traversing the heart, the myosin heads protruding from the thick filament chomp down on the nearest actin filament, then tug against it like sailors tugging in tandem on a rope, pulling the sarcomere walls closer together and making the muscle fiber contract, before relaxing again.
That’s a heartbeat. And if those myosin sailors are tugging too hard on those actin ropes? That’s hypercontractility, the hallmark of HCM.
But why would this happen?
About 35 to 40 percent of all known HCM-inducing mutations are in cardiac myosin, making it an obvious candidate for intense scrutiny. But until about a dozen years ago, nobody could study the effects of cardiac myosin mutations, Spudich said, because the biotechnology for making significant amounts of it that would work didn’t exist yet.
Then one of his frequent collaborators at the University of Colorado figured out a way to pull it off. Spudich dropped everything else he’d been doing and began systematically characterizing the effects of HCM-inducing mutations on the function of cardiac myosin molecules, asking: Why would so many diverse mutations all cause myosin to be hypercontractile?
Most of these mutations pop up on the molecule’s head. Another big batch are in the top part of its tail. Spudich hypothesized that the mutations somehow made each individual cardiac-myosin molecule faster-moving or more forceful than normal.
“But in the many mutations we studied, neither molecular speed nor strength were accounting for mutation-induced hypercontractility,” he said. “We were missing something.”
Dreaming
On Dec. 14, 2014, after many months of not getting expected results, Spudich lay awake in bed late at night wondering what clue he’d been overlooking.
“For just one night, stop thinking about your work,” Anna told him. She gave him a book she was sure he was going to like: The Haunted Mesa, by Louis L’Amour. The book’s plot unfolded in the setting of a Southwestern mesa similar to many Spudich had spied from above during his airborne sojourns in the Southwest.
He nodded off about 20 pages in, awakening hours later from a vivid dream in which the image of a mesa morphed into a myosin molecule.
It was 5:30 a.m. Aflame with inspiration, Spudich jumped out of bed and beelined to his computer, a new hypothesis in his head.
Protein molecules are born as linear sequences of chemical building blocks called amino acids, but that one-dimensionality doesn't last long. Almost as soon as each molecule gets produced in a cell, it folds up into a characteristic three-dimensional shape it will retain for the rest of its working life.
With molecular-modeling software now widely available, researchers including Spudich can speedily rotate a molecule of interest in any of three dimensions onscreen. Viewed from the right perspective, one part of the myosin head’s surface is a broad expanse, as flat as the mesa of the dream that had just awakened the sleeping scientist.
Those who study myosin have known about this mesalike surface since 1993, Spudich said. But until now, nobody had given much thought to its significance.
Every amino acid sequence of a protein molecule is specified by the gene encoding that particular protein. There are 20 different amino acid varieties to choose from, each with its own distinctive biochemical quirks: for example, a negative versus positive versus neutral electrical charge.
One typical type of mutation results in the substitution of one amino acid for another.
Spudich’s team had previously generated computer models flagging points along cardiac myosin’s linear sequence where a mutated amino acid had been found to cause HCM. These mutations’ locations seemed to be scattered pretty randomly along the molecule.
But viewed on the properly folded molecule, as Spudich was now doing, many of these mutations could be seen to fall somewhere on the mesalike surface of the head of myosin. Many others fell along the part of the tail against which the head rested when the molecule was assuming its “sleeping” position.
All hands on deck
Spudich’s group had been studying 15 or so of the most common HCM-inducing cardiac myosin mutations. They knew that most of those mutations had the effect of changing or eliminating the electrical charge the amino acid in the unmutated molecule would have had.
Now it hit him: Opposites attract. Surfaces with lots of positive charges on them are drawn to surfaces with lots of negative charges on them. Any mutation that reduces this charge opposition could result in the myosin head’s spending less time tucked up against its tail and more time on duty tugging on its nearby actin filament. These mutations aren’t changing myosin molecules’ strength or speed; they’re just making more heads available to do the tugging.
What Spudich and others had been overlooking was this: Most of the time, many of the myosin heads in the thick filament are on break — at least in healthy heart muscle. Thick fibers normally host a huge reserve army of loafing myosin heads, which is a good thing; those loafing heads can be recruited by normal physiological responses when needed. But HCM mutations effectively nudge them back on the job, even when they’re not needed.
A sarcomere’s contractile force, Spudich reasoned, is proportional to the number of myosin heads that are “grabbing” onto the actin filament at any given moment. Normally, the mesalike surface of a cardiac myosin molecule spends much of its time in coordinated proximity with a portion of the “tail” section, sequestering the head so it can’t grab the actin filament.
Looking at the folded cardiac-myosin molecule on his computer screen, Spudich realized that by weakening the overall attraction between a myosin molecule’s head and tail, HCM-inducing mutations were freeing up myosin heads to grab onto a neighboring actin filament, increasing the number of myosin heads actually pulling their weight at any one time. Hence, the hypercontractile heart. Not quite proof, but a hypothesis with a bright future.
Spudich’s group has since shown that mutations at these suspect sites do actually alter cardiac myosin’s posture and put more heads in play. A 2018 paper in Proceedings of the National Academy of Sciences co-authored by Spudich suggested that a drug called mavacamten, now in phase-3 clinical trials for HCM, may be successful in reversing the hypercontractility induced by a wide range of HCM-inducing mutations on the myosin mesa.
All patients in two earlier trials of mavacamten showed significant improvement. These trials have been sponsored by a South San Francisco-based biotechnology company, MyoKardia Inc., that Spudich co-founded in 1998 to speed the translation of his findings into drugs that could be used in clinical practice to treat HCM. Mavacamten was the fruit of this discovery effort.
“The drug is pushing available heads into the unavailable state — the opposite of what I believe most of these mutations are doing,” Spudich said.
In principle, this approach might apply to most HCM-causing mutations.
As for The Haunted Mesa, Spudich eventually finished it and, he said, enjoyed it. But the thing that sticks with him was that image of a mesa and its prophetic power, as revealed in his dream that night.
What if Spudich’s wife hadn’t given him that book to read that night? “I think someone else would have stumbled on the same idea,” he said. “The dream just jump-started it, accelerating progress by a number of years.”
For this, all hail Morpheus. “Sleep is amazing,” Spudich said. “You’ve been thinking and thinking and thinking about something for such a long time. Then suddenly the dream solves the puzzle for you.”
