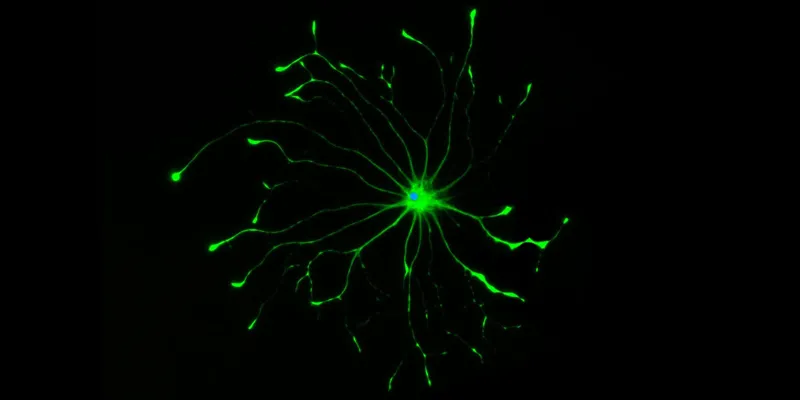
Photo courtesy Shane Liddelow: Star-shaped cells known as astrocytes are essential to the healthy function of neurons in the brain, but a new study shows that an aberrant version of astrocytes may contribute to many neurodegenerative diseases.
Stanford Medicine News Center - January 18th, 2017 - by Bruce Goldman
While most of us haven’t heard of astrocytes, these cells are four times as plentiful in the human brain as nerve cells. Now, a team led by researchers at the Stanford University School of Medicine has found that astrocytes, which perform many indispensable functions in the brain, can take on a villainous character, destroying nerve cells and likely driving many neurodegenerative diseases.
A study describing the findings was published online Jan. 18 in Nature.
“We’ve learned astrocytes aren’t always the good guys,” said the study’s senior author, Ben Barres, MD, PhD, professor of neurobiology, of developmental biology and of neurology and neurological sciences. “An aberrant version of them turns up in suspicious abundance in all the wrong places in brain-tissue samples from patients with brain injuries and major neurological disorders from Alzheimer’s and Parkinson’s to multiple sclerosis. The implications for treating these diseases are profound.”
Barres, who has spent three decades focusing on brain cells that aren’t nerve cells, called the findings “the most important discovery my lab has ever made.” Stanford postdoctoral scholar Shane Liddelow, PhD, is the study’s lead author.
Up to now, the pharmaceutical industry has mostly targeted nerve cells, also known as neurons, Barres said. But a broad range of brain disorders may be treatable by blocking astrocytes’ metamorphosis into toxic cells, or by pharmaceutically countering the neuron-killing toxin those harmful cells almost certainly secrete.
Role of astrocytes
Once thought of as mere packing peanuts whose job it was to keep neurons from jiggling when we jog, astrocytes are now understood to provide critical hands-on support and guidance to neurons, enhancing their survival and shaping the shared connections between them that define the brain’s labyrinthine circuitry. It’s also known that traumatic brain injury, stroke, infection and disease can transform benign “resting astrocytes” into “reactive astrocytes” with altered features and behaviors. But until recently, whether reactive astrocytes were up to good or evil was an open question.
In 2012, Barres and his colleagues resolved that ambiguity when they identified two distinct types of reactive astrocytes, which they called A1 and A2. In the presence of LPS, a component found in the cell walls of bacteria, they observed that resting astrocytes somehow wind up getting transformed into A1s, which are primed to produce large volumes of pro-inflammatory substances. A2s, on the other hand, are induced by oxygen deprivation in the brain, which occurs during strokes. A2s produce substances supporting neuron growth, health and survival near the stroke site.
This raised two questions: How are A1s generated? And once they’re generated, what do they do? The new study answers both questions.
In addressing the first question, the study showed that the brain’s immune cells, microglia, which are known to become activated by LPS exposure as well as in most brain injuries and diseases, begin spewing out pro-inflammatory factors that change astrocytes’ behavior.
Pro-inflammatory factors
In a series of experiments using laboratory mice, the scientists identified three pro-inflammatory factors whose production was ramped up after LPS exposure: TNF-alpha, IL-1-alpha and C1q. In the brain, all three of these substances are secreted exclusively by microglia. Each, by itself, had a partial A1-inducing effect on resting astrocytes. Combined, they propelled resting astrocytes into a full-fledged A1 state.
Next, the researchers confirmed that A1s jettison the nurturing qualities they’d had as resting astrocytes, which Barres’ group has shown are essential to the formation and functioning of synapses, and instead became toxic to neurons.
In vertebrates, nerve cells called retinal ganglion cells send information from the retina to vision-processing centers in the brain. RGCs can thrive in culture, but only if accompanied by astrocytes. The scientists cultured rodent RGCs with either resting or A1 astrocytes and counted the resulting synapse numbers. RGCs cultured in combination with A1s produced only half as many synapses as RGCs grown with resting astrocytes, and those that formed didn’t work very well.
Further experiments showed that A1s lose resting astrocytes’ capacity to prune synapses that are no longer needed or no longer functional and whose continued existence undermines efficient brain function.
Indeed, when the researchers cultured healthy RGCs with increasingly stronger concentrations of the broth in which A1s had been bathing, almost all the RGCs eventually died. This and other experiments showed that A1s secrete a powerful, neuron-killing toxin.
The same treatment killed many other types of neurons, including both the spinal motor neurons that die in amyotrophic lateral sclerosis and the human dopaminergic neurons whose mysterious loss is the cause of Parkinson’s disease. A1 bathwater also impaired the development of yet another class of non-neuron brain cells called oligodendrocytes — essentially fat-filled flapjacks that wrap themselves around nerve fibers, providing electrical insulation that speeds long-distance signal propagation. Autoimmune destruction of oligodendrocytes and their fatty contents gives rise to multiple sclerosis.
Staving off A1 formation
In another experiment, the researchers severed rodents’ optic nerves — an act ordinarily lethal to RGCs, whose outgoing fibers, called axons, constitute the optic nerve. In the central nervous system, severing axons causes the entire neuron to die quickly, but why they die has been a mystery. The investigators determined the cause: A1s. They observed that those reactive astrocytes formed quickly after axons were severed, but that neutralizing TNF-alpha, IL-1-alpha and C1q with antibodies to these three substances prevented A1 formation and RGC death in the animals.
Finally, the researchers analyzed samples of human brain tissue from patients with Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis and multiple sclerosis. In every case, they observed large numbers of A1s preferentially clustering where the disease was most active. For example, in the samples from Alzheimer’s patients, nearly 60 percent of the astrocytes present in the prefrontal cortex, a region where the disease takes a great toll, were of the A1 variety. Because A1s are highly toxic to both neurons and oligodendrocytes, these findings strongly imply that A1 formation is helping to drive neurodegeneration in these diseases.
An effort to identify the neurotoxin secreted by A1 astrocytes is underway, Barres said. “We’re very excited by the discovery of neurotoxic reactive astrocytes,” he said, “because our findings imply that acute injuries of the retina, brain and spinal cord and neurodegenerative diseases may all be much more highly treatable than has been thought.”
The work is an example of Stanford Medicine’s focus on precision health, the goal of which is to anticipate and prevent disease in the healthy and precisely diagnose and treat disease in the ill.
Barres is the co-founder of a biotechnology company, Annexon Biosciences, which has produced, and filed for a patent for, an inhibitory antibody to C1q. Drugs to block TNF-alpha and IL1-alpha already exist.
Other Stanford co-authors are graduate student Kevin Guttenplan; postdoctoral scholars Laura Clarke, PhD, Todd Peterson, PhD, Brooke Napier, PhD, and Christopher Bohlen, PhD; Frederick Bennett, MD, an instructor of psychiatry and behavioral sciences; medical student Mariko Bennett, PhD; life science research assistant Alexandra Munch; former postdoctoral scholar Won-Suk Chung, PhD; and Marion Buckwalter, MD, PhD, associate professor of neurosurgery and of neurology and neurological sciences.
Researchers from the University of California-San Francisco, the Technical University of Munich, Boston Children’s Hospital, Johns Hopkins University and Harvard University also are co-authors.
The study was funded by the National Institutes of Health (grants R01AG048814 and R01DA015043), the Christopher and Dana Reeve Foundation, the Novartis Institute for Biomedical Research, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the JPB Foundation, the Cure Alzheimer’s Fund, the Glenn Foundation and Vincent and Stella Coates.
Stanford’s Department of Neurobiology also supported the work.
