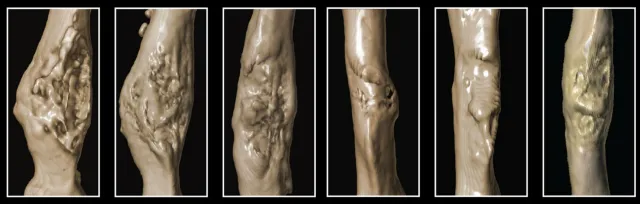
Graphic courtesy Tevlin et al: The bones of diabetic mice are more fragile than those of control animals when they heal
after a fracture. Bones of the control mice (left) form larger calluses during healing, making the repairs stronger. In
contrast, the bones of the diabetic mice (right) have smaller calluses, making the repairs more brittle.
Stanford Medicine News Center - January 11th, 2017 - by Krista Conger
Bone fractures in diabetic mice heal better in the presence of a protein that stimulates the activity of skeletal stem cells, according to a study by researchers at the Stanford University School of Medicine.
The protein counteracts a decrease in stem cell activity that the researchers observed both in mouse models of diabetes and in bone samples from diabetic patients who had undergone joint replacements. The researchers hope the discovery will lead to ways to help people with diabetes heal more efficiently from broken bones.
“We’ve uncovered the reason why some patients with diabetes don’t heal well from fractures, and we’ve come up with a solution that can be locally applied during surgery to repair the break,” said Michael Longaker, MD, co-director of Stanford’s Institute for Stem Cell Biology and Regenerative Medicine. “Diabetes is rampant worldwide, and any improvement in the ability of affected people to heal from fractures could have an enormously positive effect on their quality of life.”
The study was published Jan. 11 in Science Translational Medicine. Longaker, a professor of plastic and reconstructive surgery, shares senior authorship of the study with Charles Chan, PhD, an instructor at the stem cell institute. Postdoctoral scholar Ruth Tevlin, MD, is the lead author.
Healing difficulties
Diabetes mellitus is a metabolic disease characterized by the inability to either produce or to respond appropriately to insulin. It affects hundreds of millions of people worldwide and is increasing in prevalence. In addition to causing dangerous swings in blood sugar levels after meals, the condition leads to many other debilitating symptoms, including an impaired ability to heal soft tissue injuries and skeletal fractures. The precise molecular reason behind this impaired bone healing has been unknown, however.
Longaker, Chan and Tevlin built on previous research in which they and colleagues in the laboratory of co-author Irving Weissman, MD, professor of pathology and of developmental biology, identified and described a population of cells in the bones of mice that serve as skeletal stem cells, or SSCs. These adult stem cells can become all components of the skeletal system, including bone, cartilage and a part of the bone marrow known as the stroma. They subsequently showed that fracture healing in mice was severely impaired when these stem cells were depleted. That finding got them thinking.
“We wanted to apply what we knew about skeletal stem cells to the problem of impaired bone healing in people with diabetes,” said Chan. “Does the disease affect fracture healing by somehow modulating the activity of these stem cells?”
The researchers used a mouse model of Type 2 diabetes, in which the disease arises when the animals are about 4 weeks old. Prior to the development of the disease, the prediabetic mice were able to heal leg bone fractures as effectively as wild-type mice, the researchers found. In contrast, after the disease had manifested itself, the repaired bone was significantly weaker and less dense than the bone in the control animals. When they compared the numbers of SSCs in the healing bone seven days after fracture, they found that the diabetic mice had significantly lower numbers of these cells than did the control animals.
Signaling problem
A series of experiments ruled out a systemic reason for this reduction in stem cell numbers, and also confirmed that the cells themselves were fully functional. That left only a potential problem with the signals the cells were receiving from the surrounding environment, or niche. When Tevlin and her colleagues analyzed that environment, they found that the diabetic animals produced significantly lower levels of a family of signaling proteins, called hedgehog, that is known to play a critical role in many biological processes, including embryonic development and tissue regeneration.
The researchers collaborated with co-author < target="_blank"="https://med.stanford.edu/profiles/philip-beachy">Philip Beachy, PhD, professor of biochemistry and of developmental biology, to test whether artificially blocking the hedgehog signaling pathway could impair bone healing in nondiabetic mice. They found that control mice exposed to a molecule that blocked the pathway regrew bone that was weaker and more brittle — just like the diabetic animals.
“Next we had to test whether adding the hedgehog signaling proteins back into the local environment in diabetic animals restored their ability to heal fractures,” said Longaker. The researchers collaborated with co-authors < target="_blank"="https://med.stanford.edu/profiles/fan-yang">Fan Yang, PhD, assistant professor of bioengineering and orthopaedic surgery, and postdoctoral scholar Xinming Tong, PhD, to devise a biologically friendly hydrogel into which the hedgehog signaling proteins were embedded. The gel was applied directly to the fracture site. “And these animals healed just like normal mice,” said Longaker, who holds the Deane P. and Louise Mitchell Professorship in the School of Medicine.
Clues in human bone samples
Finally, the team reached out to co-author Stuart Goodman, MD, PhD, professor of orthopaedic surgery, to obtain bone samples from patients with diabetes who were undergoing joint replacement for osteoarthritis. They compared the expression of proteins important to the hedgehog signaling pathway from these samples with others obtained from non-diabetic patients. Normally this tissue would be discarded by the surgeon, but in this case it held important clues.
“What we saw in these human samples completely echoed what we saw in the mice,” said Chan. “The bones from the diabetic patients displayed significantly reduced expression of these important signaling proteins.”
Longaker, Chan and Tevlin believe the inhibition of the hedgehog signaling pathway arises from diabetes-associated inflammation that causes high levels of a molecule called tumor necrosis factor alpha. TNF-alpha levels are known to be elevated in patients with diabetes, and the researchers observed a corresponding increase in their mouse models of the disease. They also showed that these increased levels of TNF-alpha inhibited the expression of some hedgehog family members. Directly inhibiting all TNF-alpha activity, however, could have other dire consequences for an animal or a human patient because TNF-alpha plays many important biological roles.
“Here we’ve devised a feasible strategy for reversing a tissue-specific pathology — the inability to heal skeletal fractures efficiently — in a complex metabolic disease like diabetes, through the local application of a compound to stimulate the activity of adult stem cells,” said Longaker said. “We anticipate that hedgehog-mediated molecular therapies that directly target stem cells in human patients could be therapeutic.”
More research is necessary before trying this approach in humans, but the researchers are hopeful that local application of hedgehog proteins will be shown to be both safe and effective. Their findings further validate the idea that tissue-specific stem cells are likely to play vital roles in tissue regeneration and response to injury.
“This research represents a significant step forward toward realizing the promise of Proposition 71, which established the California Institute for Regenerative Medicine,” said Chan. “We’ve looked to stem cells to learn why people with diabetes don’t heal bone fractures properly, and come up with an approach that we are excited to try in the clinic.”
The paper provides a complete list of the Stanford co-authors of the study.
The research was supported by the National Institutes of Health (grants R56DE025597, R01DE021683, R21DE024230, R01DE019434, RC2DE020771, U01HL099776, R21DE019274, U01HL099999, 5R01CA86065, 5 R01L058770 and R01DE024772), the Thomas and Stacey Siebel Foundation, the Prostate Cancer Foundation, the National Institute on Aging, the California Institute for Regenerative Medicine, the Oak Foundation, the Hagey Laboratory for Pediatric Regenerative Medicine, the Gunn/Olivier Research Fund, the National Science Foundation, the Stanford University Transplant and Tissue Engineering Center, the Plastic Surgery Foundation/Plastic Surgery Research Council and the American Society of Maxillofacial Surgeons.
Stanford’s Department of Surgery also supported the work.




